
























































































































































Model Organisms
Overview
The Model Organisms Screening Center (MOSC) for the Undiagnosed Diseases Network (UDN) is composed of two Centers that use fruit fly (Drosophila melanogaster), nematode worm (Caenorhabditis elegans) and zebrafish (Danio rerio) genetics and biology to tackle rare and undiagnosed diseases. By combining state-of-the-art genetic and genomic technologies, the MOSCs investigate whether a rare variant identified in the genomes of UDN participants may contribute to disease pathogenesis. The Baylor College of Medicine (BCM)-University of Oregon (UO) MOSC is led by Hugo J. Bellen (BCM), Michael F. Wangler (BCM), Shinya Yamamoto (BCM), Monte Westerfield (UO), and John Postlethwait (UO). The Washington University in St. Louis (WashU) MOSC is led by Lilianna Solnica-Krezel, Tim Schedl, Dustin Baldridge, and Stephen C. Pak.
BCM-UO MOSC Leadership
-

- Hugo J. Bellen, DVM, PhD (PD/PI)

-
- Michael F. Wangler, MD

-
- Shinya Yamamoto, DVM, PhD

-
- Monte Westerfield, PhD

-
- John Postlethwait, PhD

WashU MOSC Leadership
-
- Lilianna Solnica-Krezel, PhD (PD/PI)

-
- Tim Schedl, PhD (PD/PI)

-
- Dustin Baldridge, MD, PhD

-
- Stephen C. Pak, PhD

Why flies, worms and zebrafish?
Over the past century, genetic model organisms have taught us so much about human biology and disease mechanisms. Although these organisms (e.g. bacteria, yeast, worm, fly, zebrafish, mouse) may look very different from us, fundamental biological mechanisms and genes are well conserved throughout evolution. To investigate the functional consequences of hundreds of rare variants found through sequencing UDN participants’ and their family members’ genomes, the MOSCs use three model organisms, fruit fly (Drosophila melanogaster), nematode worm (Caenorhabditis elegans) and zebrafish (Danio rerio). These animals are cost efficient, have short life-cycles and are amenable to sophisticated genetic manipulations to “model” a human disease condition. Drosophila, C. elegans and zebrafish are complementary to one another, providing synergistic strengths. Candidate genes and variants that are shown to have functional impacts can be further pursued in mammalian model systems, such as mouse and human pluripotent stem cells, for further translational studies.
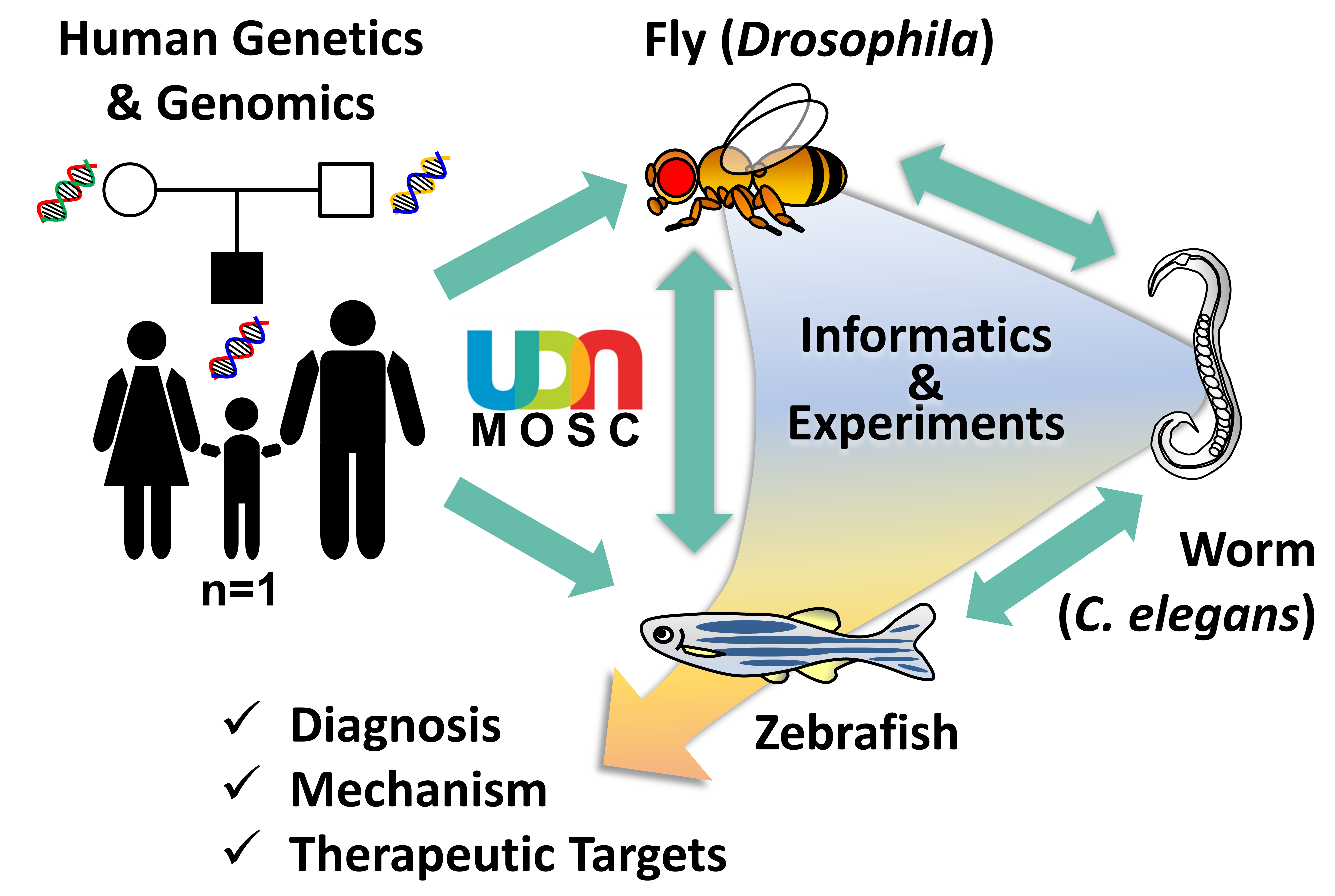
Workflow
When a diagnosis is not reached after performing a thorough clinical, genetic and/or metabolomic workup, the UDN Clinical Sites submit candidate gene(s)/variant(s) to the MOSCs together with a brief description of the participant’s condition. The MOSCs then perform database searches using a number of bioinformatics tools, including the MARRVEL tool (marrvel.org, see below), to aggregate existing information on the human gene/variant and its model organism orthologs. The MOSCs also try to identify other individuals with similar genotype and phenotype in other cohorts, a practice known as “matchmaking”. Once a variant is considered to be a high priority candidate, experiments to assess gene and variant function are designed by MOSC investigators and pursued in the C. elegans Core, Drosophila Core or Zebrafish Cores.

MARRVEL
In collaboration with Drs. Zhandong Liu’s (BCM) and Norbert Perrimon’s (Harvard Medical School) bioinformatics team, the BCM-UO MOSC developed a powerful online tool that allows anyone to quickly gather gene and variant function information. MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration) is a novel web-based tool that integrates human and model organism databases to facilitate molecular diagnosis. MARRVEL can also be used by model organism researchers to assess whether specific model organism genes of interest may have links to human diseases. MARRVEL is publicly available for clinicians and researchers worldwide at marrvel.org, and we will be continuously updating and upgrading this tool for the community.
Publications
Research Articles
AI-MARRVEL – A Knowledge-Driven AI System for Diagnosing Mendelian Disorders
Loss of the endoplasmic reticulum protein Tmem208 affects cell polarity, development, and viability
Macrocephaly and developmental delay caused by missense variants in RAB5C
Bi-allelic variants in INTS11 are associated with a complex neurological disorder
SPTSSA variants alter sphingolipid synthesis and cause a complex hereditary spastic paraplegia
The recurrent de novo c.2011C>T missense variant in MTSS2 causes syndromic intellectual disability
The microRNA processor DROSHA is a candidate gene for a severe progressive neurological disorder
Loss- or Gain-of-Function Mutations in ACOX1 Cause Axonal Loss via Different Mechanisms
IRF2BPL Is Associated with Neurological Phenotypes
Biallelic Mutations in ATP5F1D, which Encodes a Subunit of ATP Synthase, Cause a Metabolic Disorder
Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially
A Syndromic Neurodevelopmental Disorder Caused by De Novo Variants in EBF3
Tutorial Articles
Using MARRVEL v1.2 for Bioinformatics Analysis of Human Genes and Variant Pathogenicity
In Vivo Functional Study of Disease-associated Rare Human Variants Using Drosophila
Review Articles
Integrating non-mammalian model organisms in the diagnosis of rare genetic diseases in humans
Sphingolipids in neurodegenerative diseases
Drosophila as a diet discovery tool for treating amino acid disorders
‘Fly-ing’ from rare to common neurodegenerative disease mechanisms
Using Drosophila to drive the diagnosis and understand the mechanisms of rare human diseases.
Fruit flies in biomedical research
Morgan’s legacy: fruit flies and the functional annotation of conserved genes
Bedside Back to Bench: Building Bridges between Basic and Clinical Genomic Research
Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research
Funding
Photos
-
- C. elegans Core at Washington University in St. Louis

-
- C. elegans stained with a DNA dye

-
- Drosophila Core at Baylor College of Medicine

-
- Vial of transgenic fruit flies

-
- Zebrafish Core at the University of Oregon

-
- Zebrafish Core at Washington University in St. Louis
