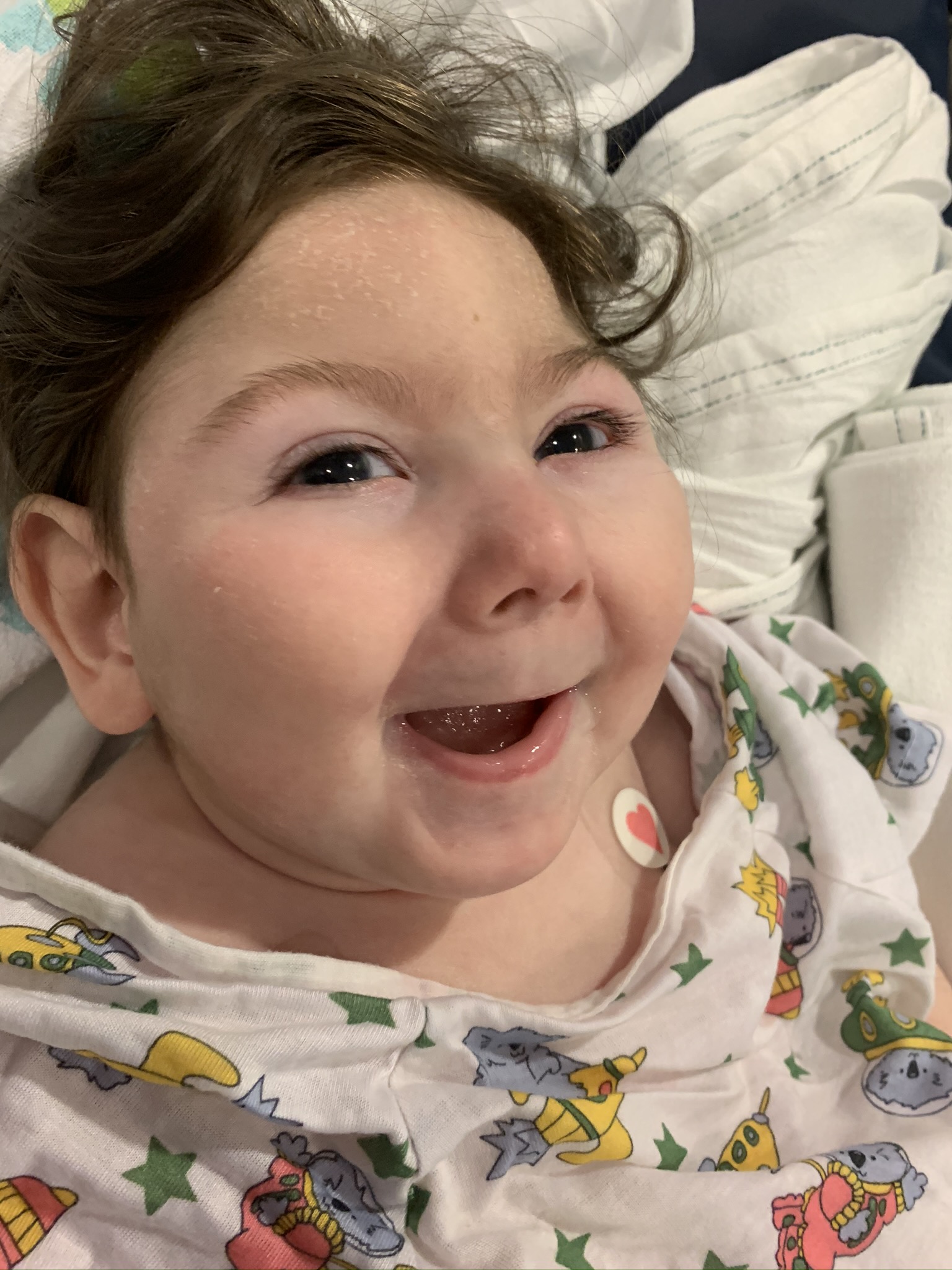
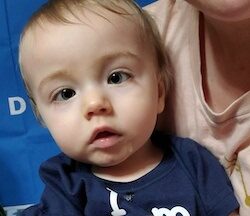
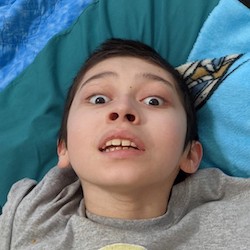
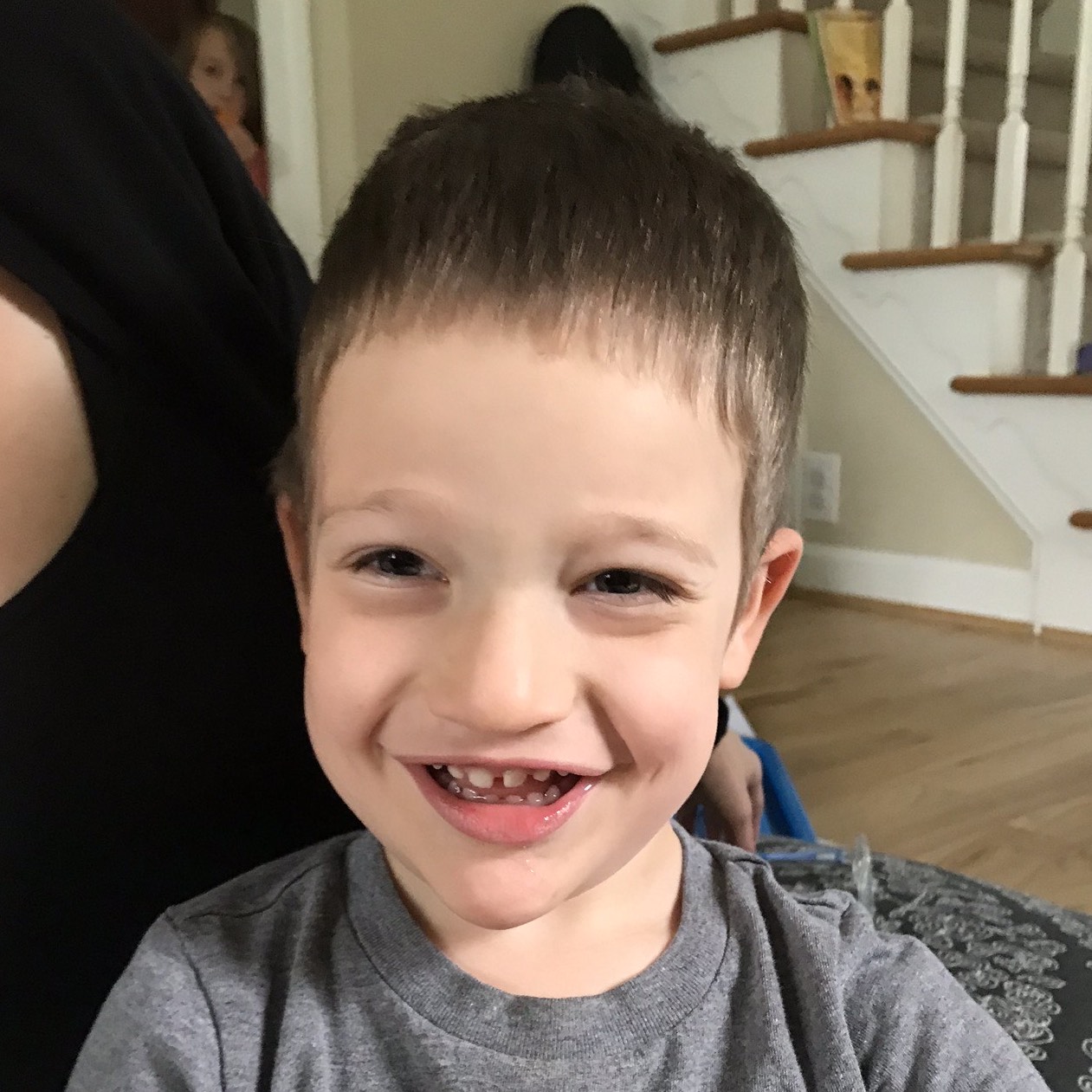
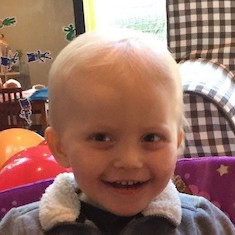
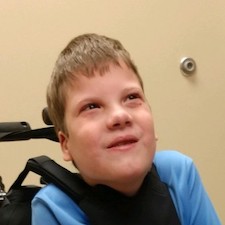
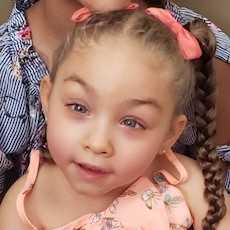
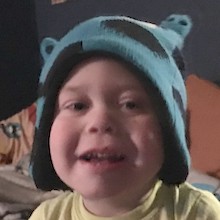
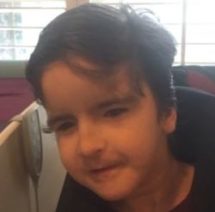
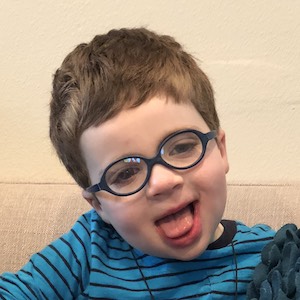
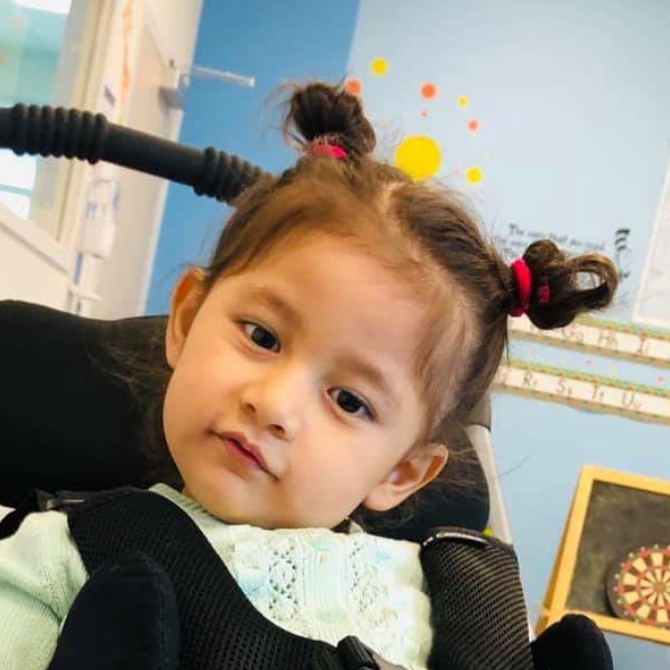
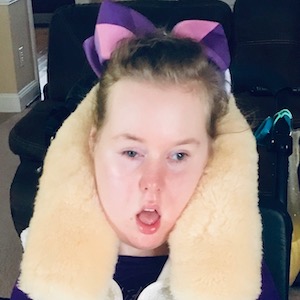
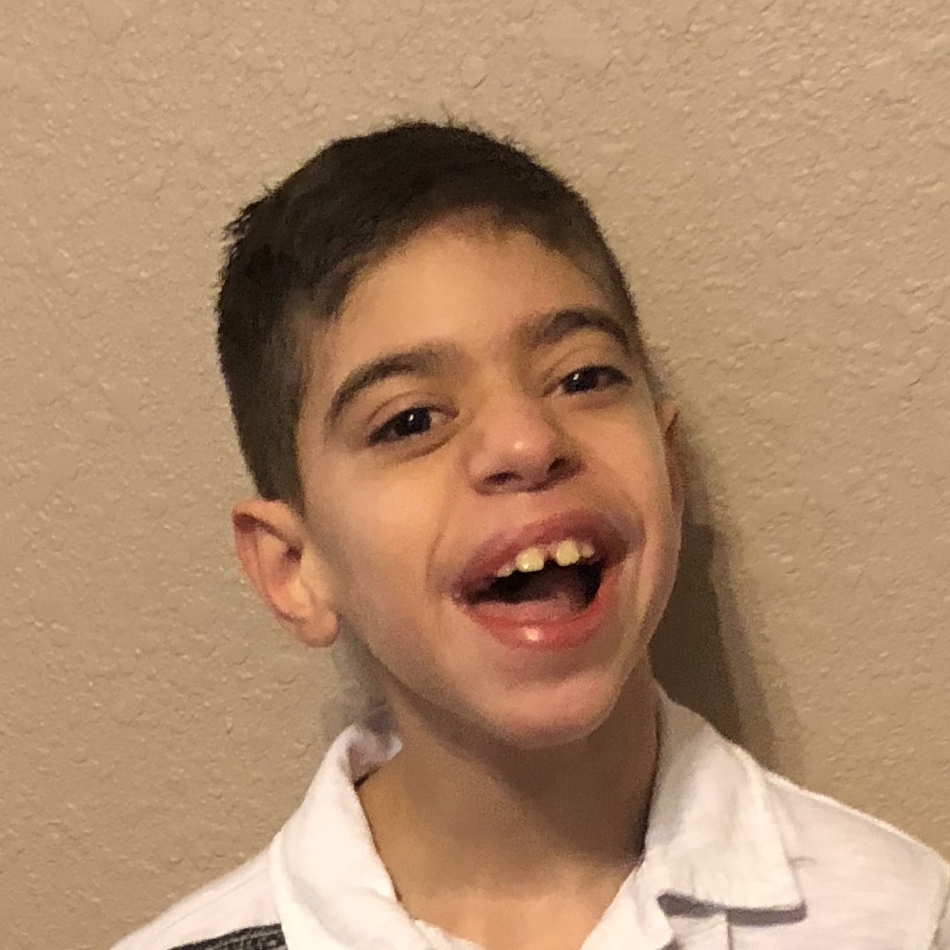
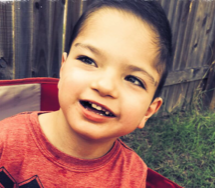
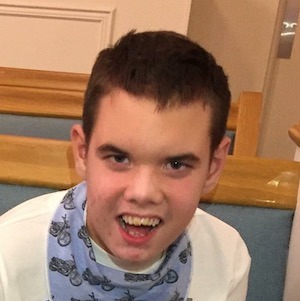
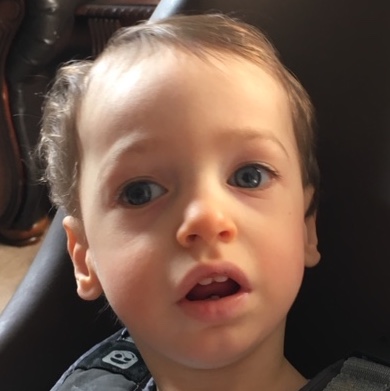
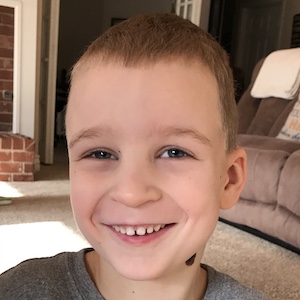
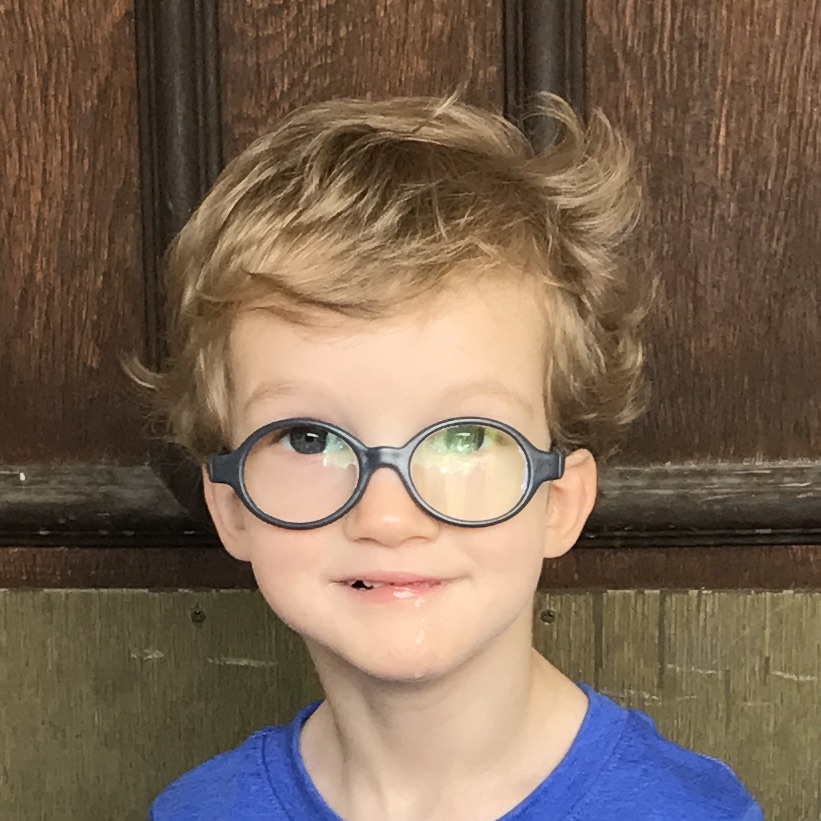
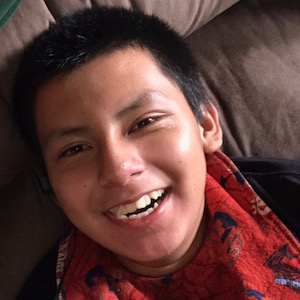
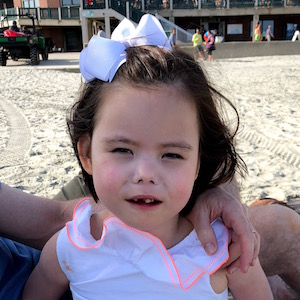
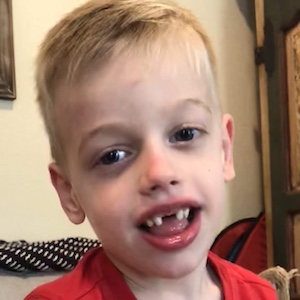
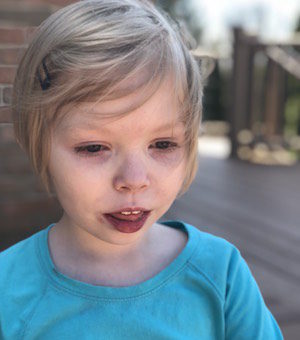
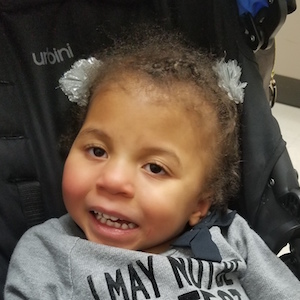
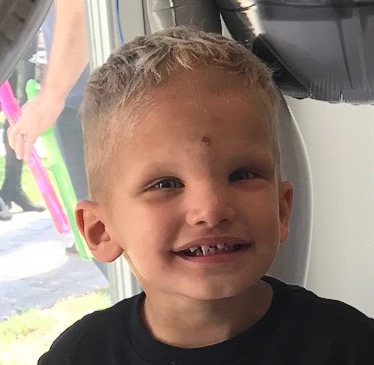
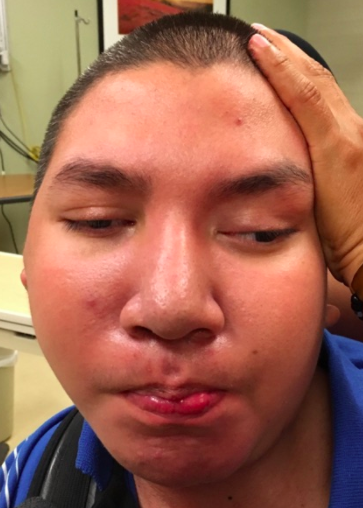
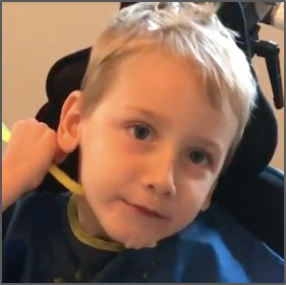
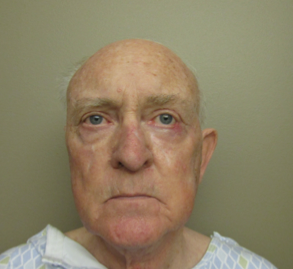
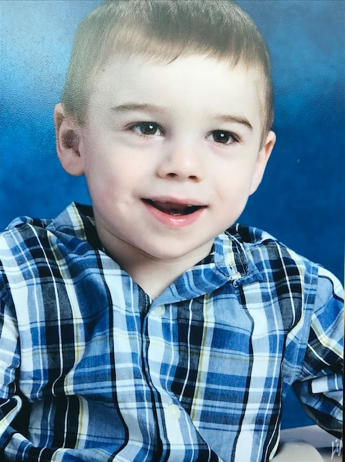
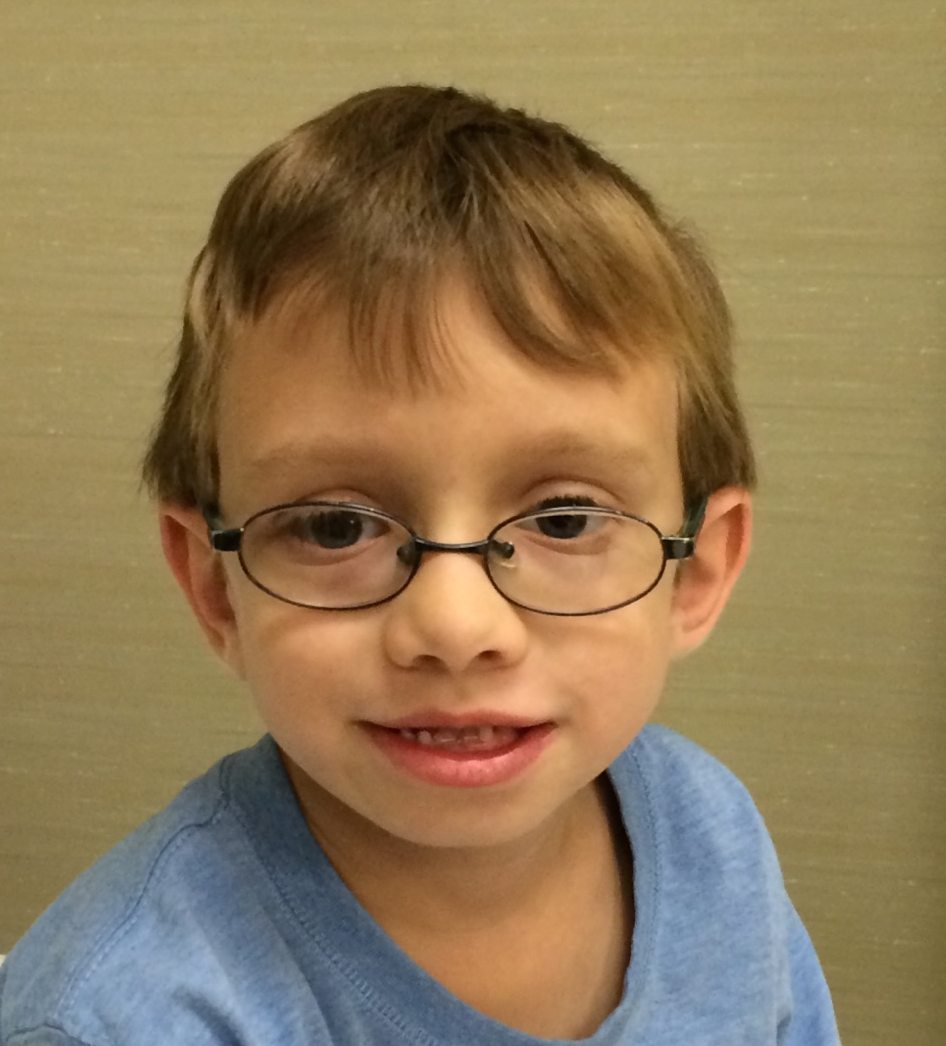
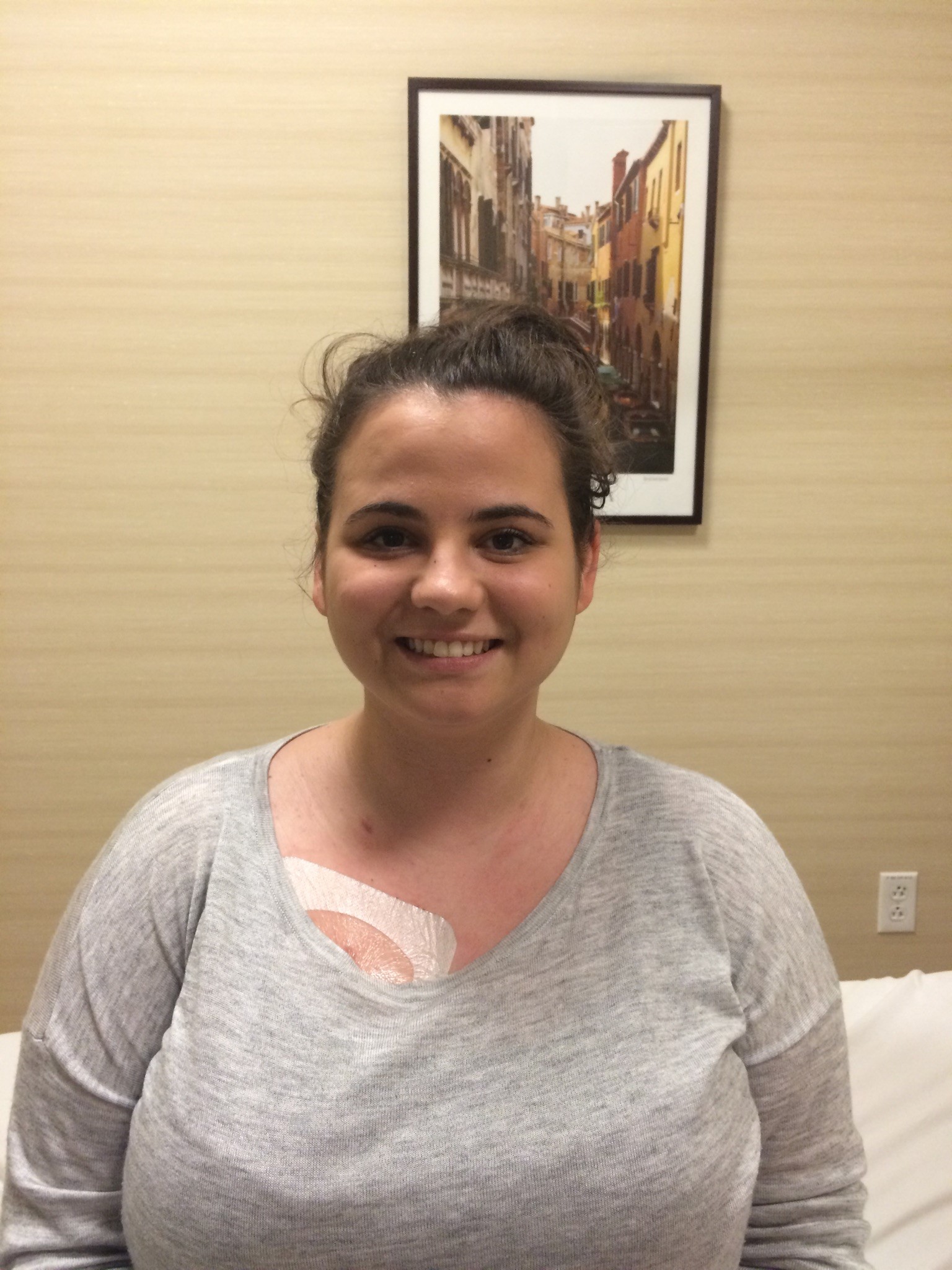
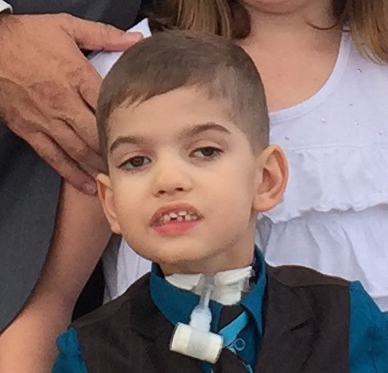
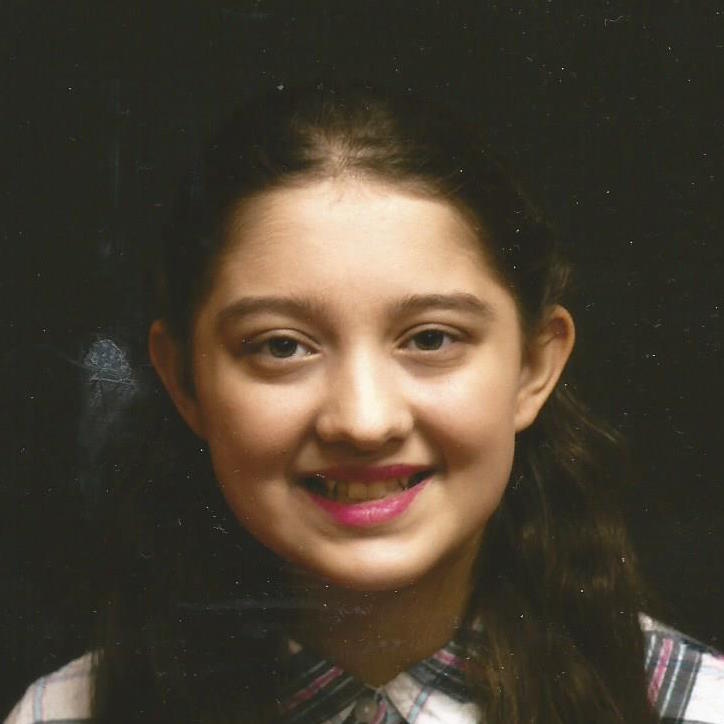Male, age 11, with gross motor delay, language impairment (aphasia), and autism
Dec 08, 2023
The participant was born at full-term. He was delivered by C-section because he had stopped moving down the birth canal (arrest of descent).
At 9 months, his parents become concerned when he was not crawling or pulling up to stand. At 15 months of age, he began interventional therapy with infant stimulation and physical therapy. At 19 months of age, he started crawling. He did not stand up until 2 years of age. He was diagnosed with having autism spectrum disorder at 3 ½ years of age.
At 4 years of age, doctors thought the participant may have an Angelman syndrome-like disorder. He was treated on a therapeutic trial of Sinemet. Within a year, he began walking. The treatment was later discontinued because it was no longer beneficial to the participant.
The participant began experiencing seizures at 7 years of age. He also has a long history of poor appetite, diarrhea, and constipation. He has flexible hips, which is likely from sitting with his feet externally rotated on the floor.
The participant has a previous diagnosis of cerebral palsy and there is question of dopa-responsive dystonia.
The participant has a younger sister with similar symptoms. Her developmental delays began at 12 months. She is also nonverbal with motor delays. She was also trialed on Sinemet but did not tolerate it.
Clinicians and researchers are investigating the following genetic change to see if it is causing the participant’s symptoms:
Phase: FAT1 and DST variants are known to be biallelic / in trans.
Segregation: FAT1 and DST variants segregate with similarly affected sister in compound heterozygous state.
Participant’s sister is also heterozygous for the CAMTA1 variant (maternally inherited).
If this participant sounds like you or someone you know, please contact us!