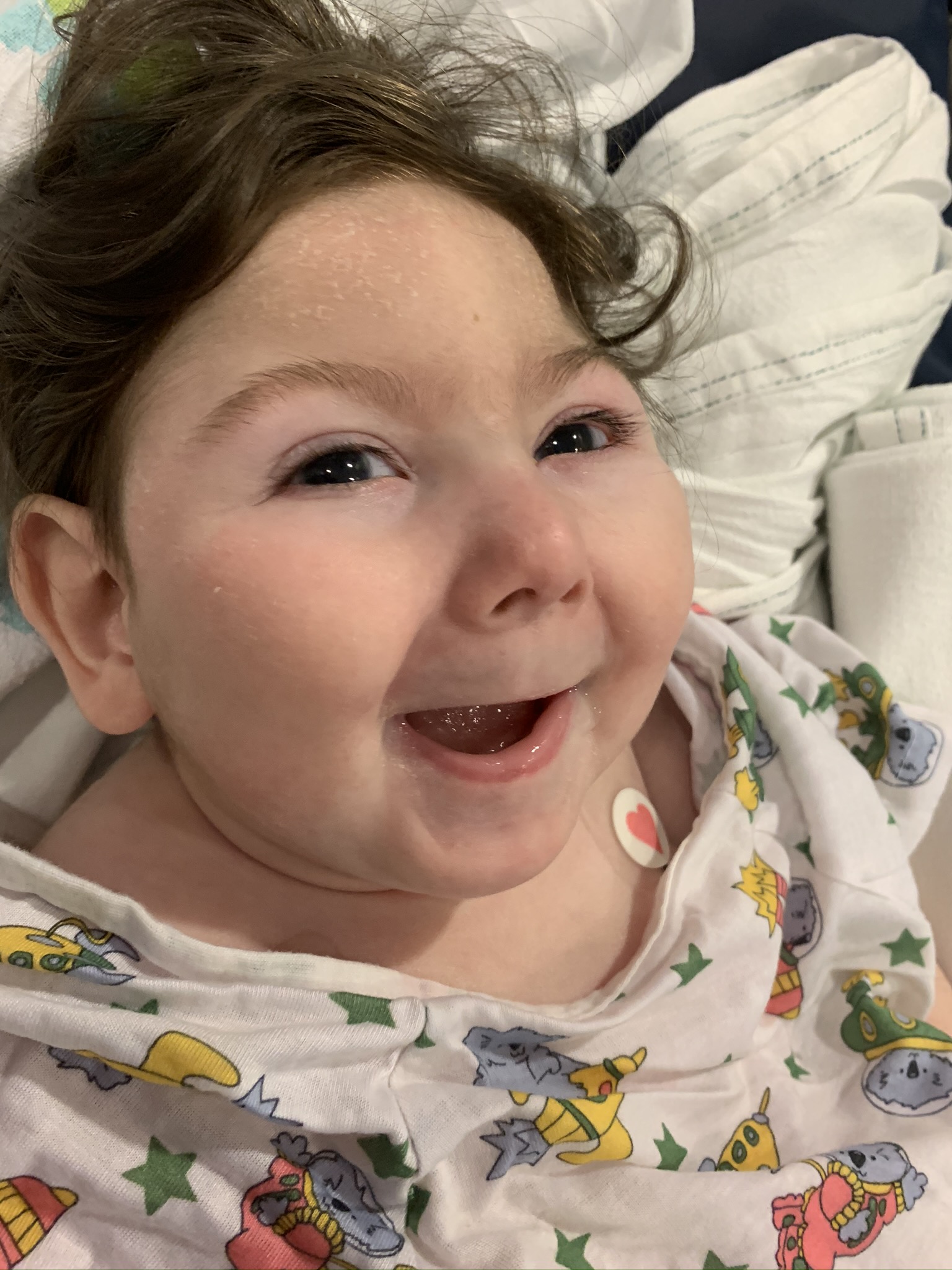
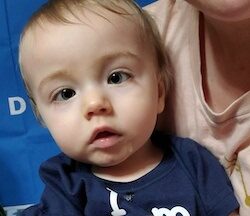
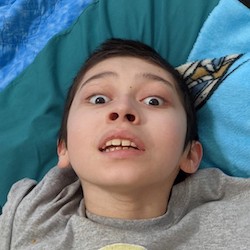
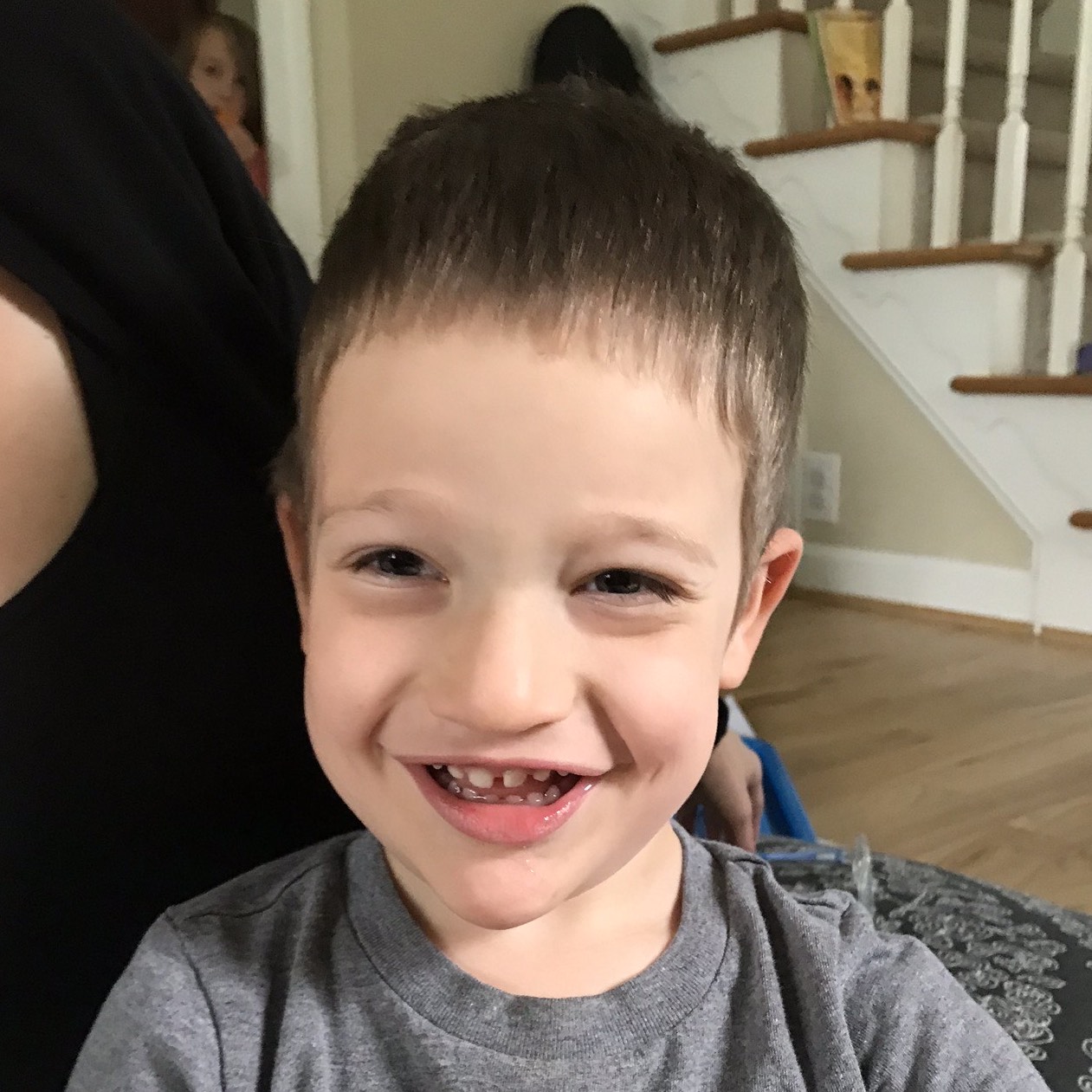
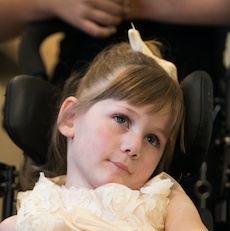
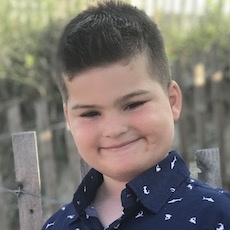
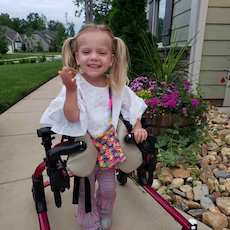
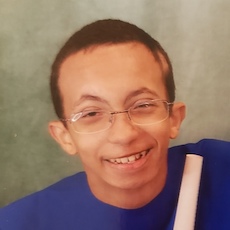
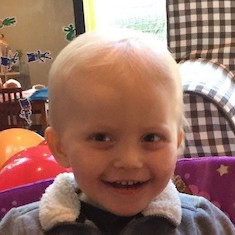
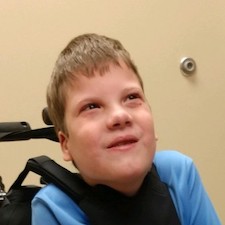
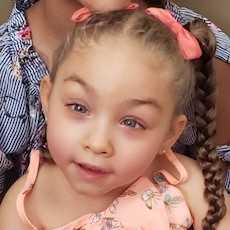
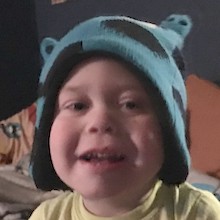
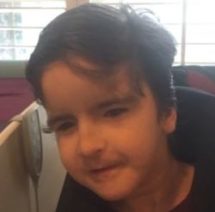
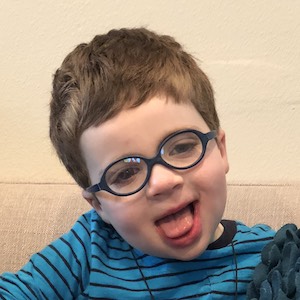
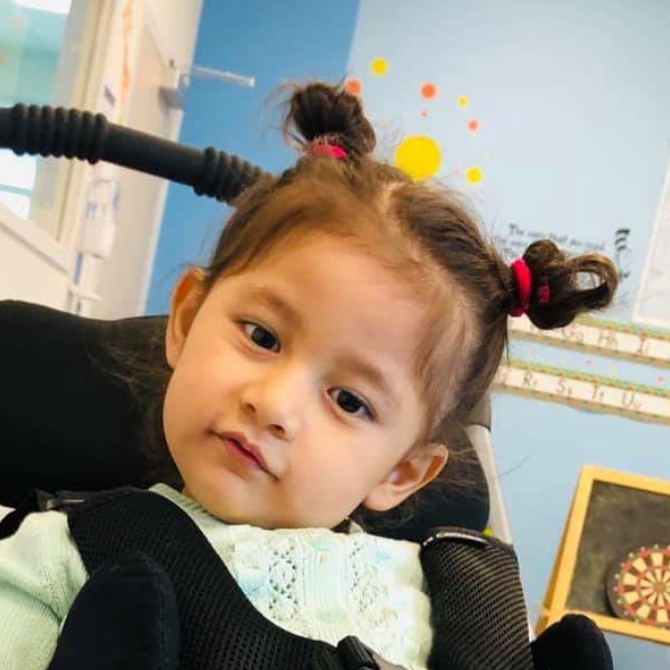
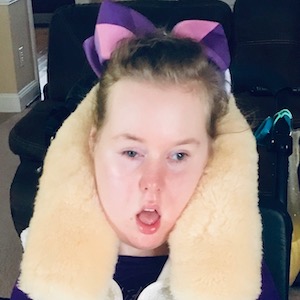
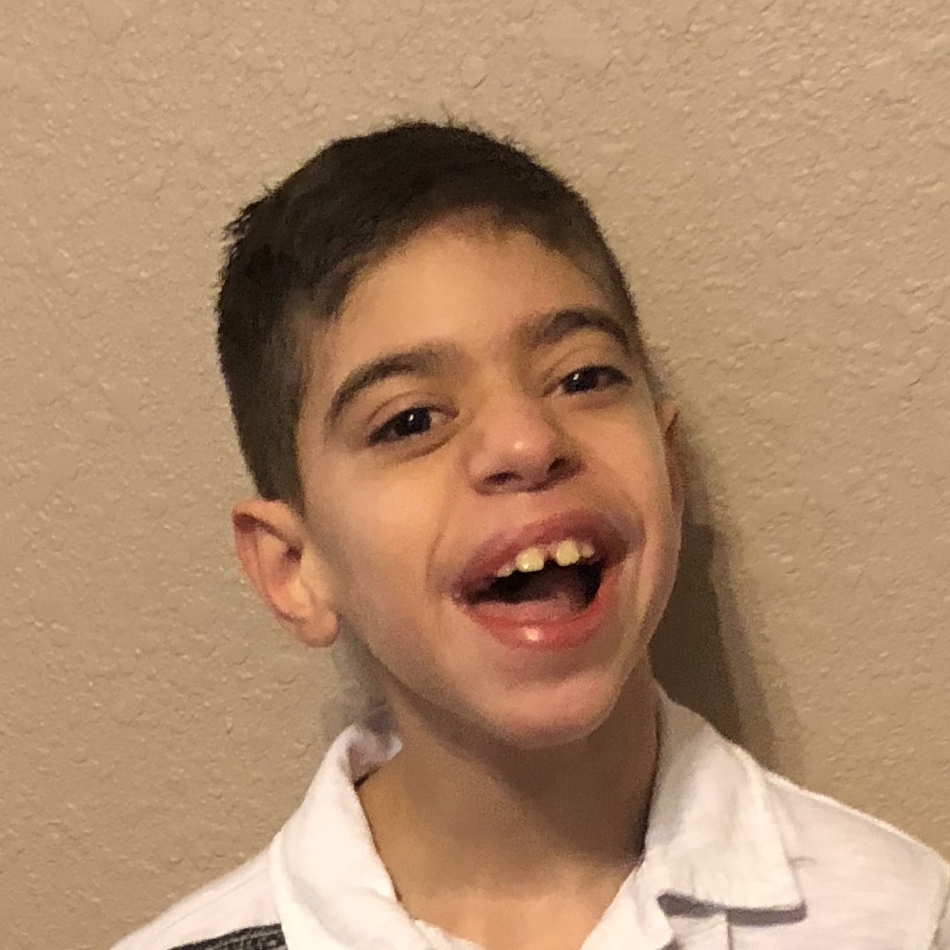
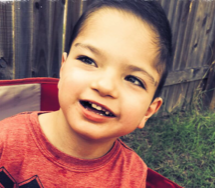
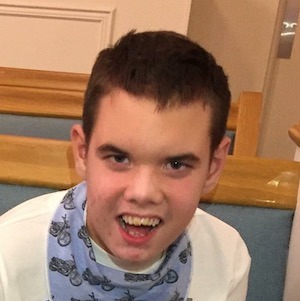
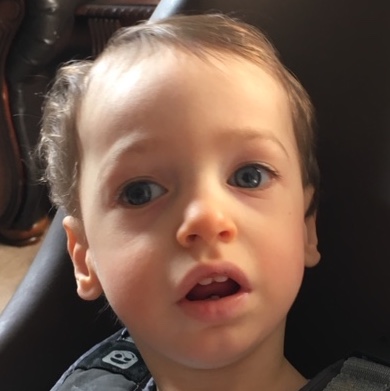
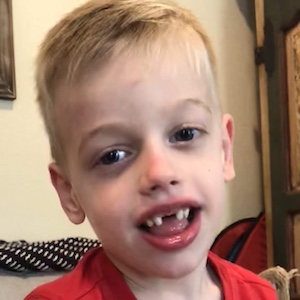
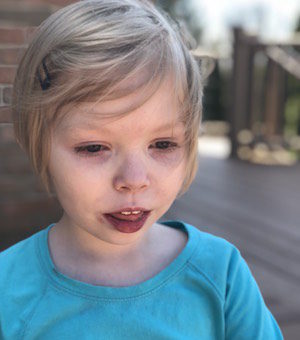
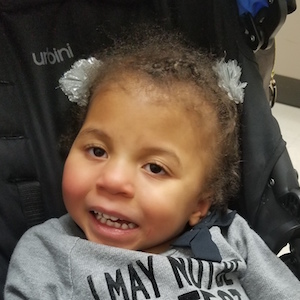
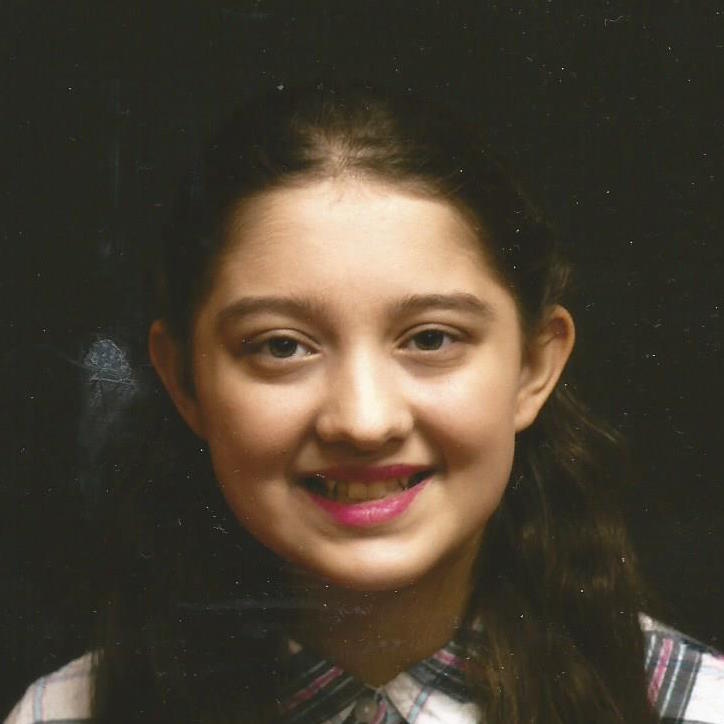Female, age 11, with Wieacker-Wolff syndrome
May 14, 2018
At 7 months, the participant’s parents noticed that she was not sitting or rolling. By 18 months, she was only babbling and crawling on her stomach. At the age of 2, the participant began to have seizures, which were sometimes associated with fevers.
Currently the participant attempts to speak, but doesn’t have any true words. She uses an iPad to communicate. She needs a wheelchair, but her leg strength has been improving slowly with physical therapy.
The participant also has low tone (hypotonia) and distinct facial features (small jaw (micrognathia), rounded nose (bulbous nose), low-set ears, upslanting eyes (palpebral fissure)).
Clinicians and researchers have identified the following genetic change to be causing the patient’s symptoms:
Xq11.2 deletion (base pair coordinates (hg19): 64,171,841-64,267,316, gene involved: ZC4H2)
If this participant sounds like you or someone you know, please contact us!