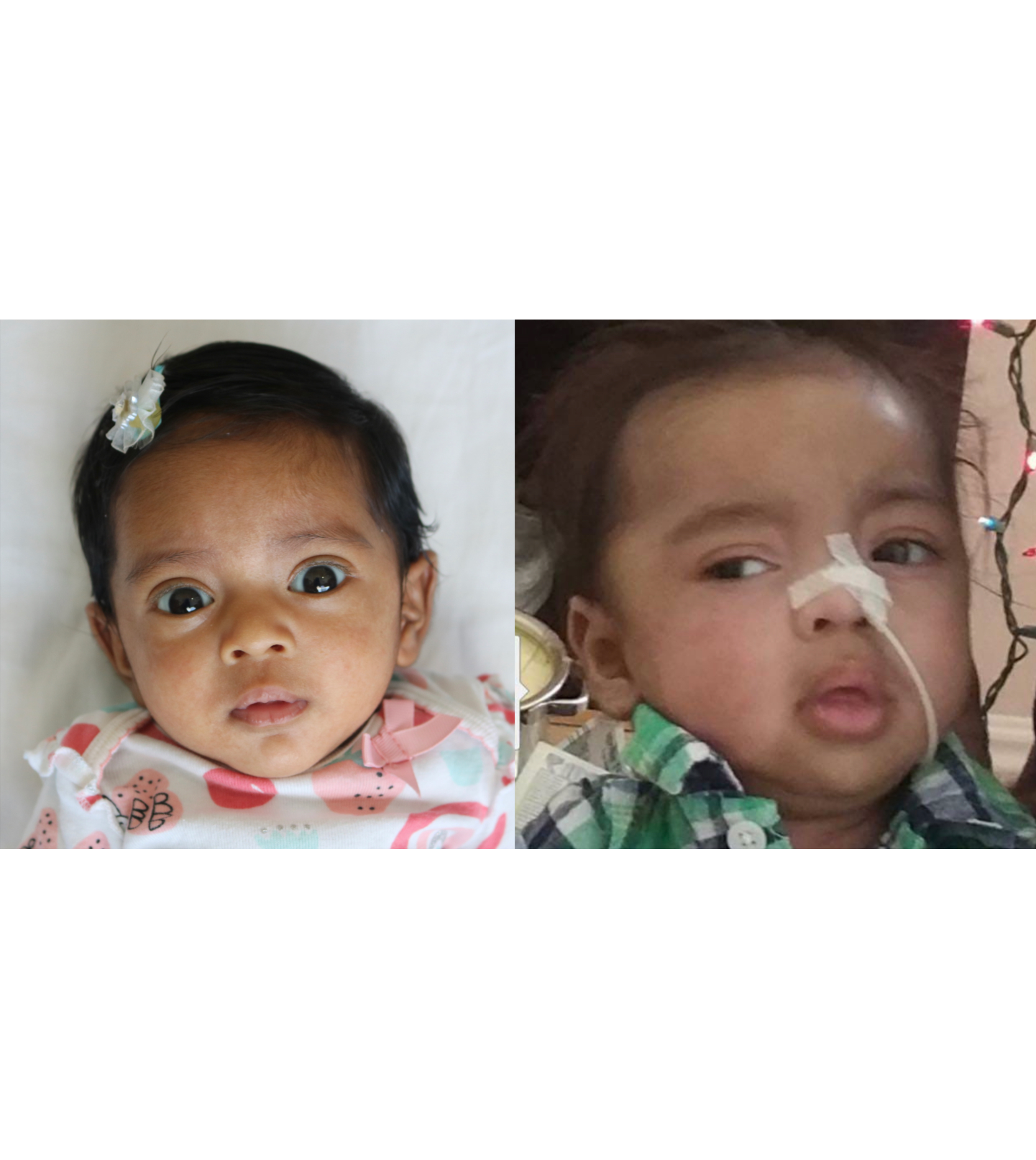
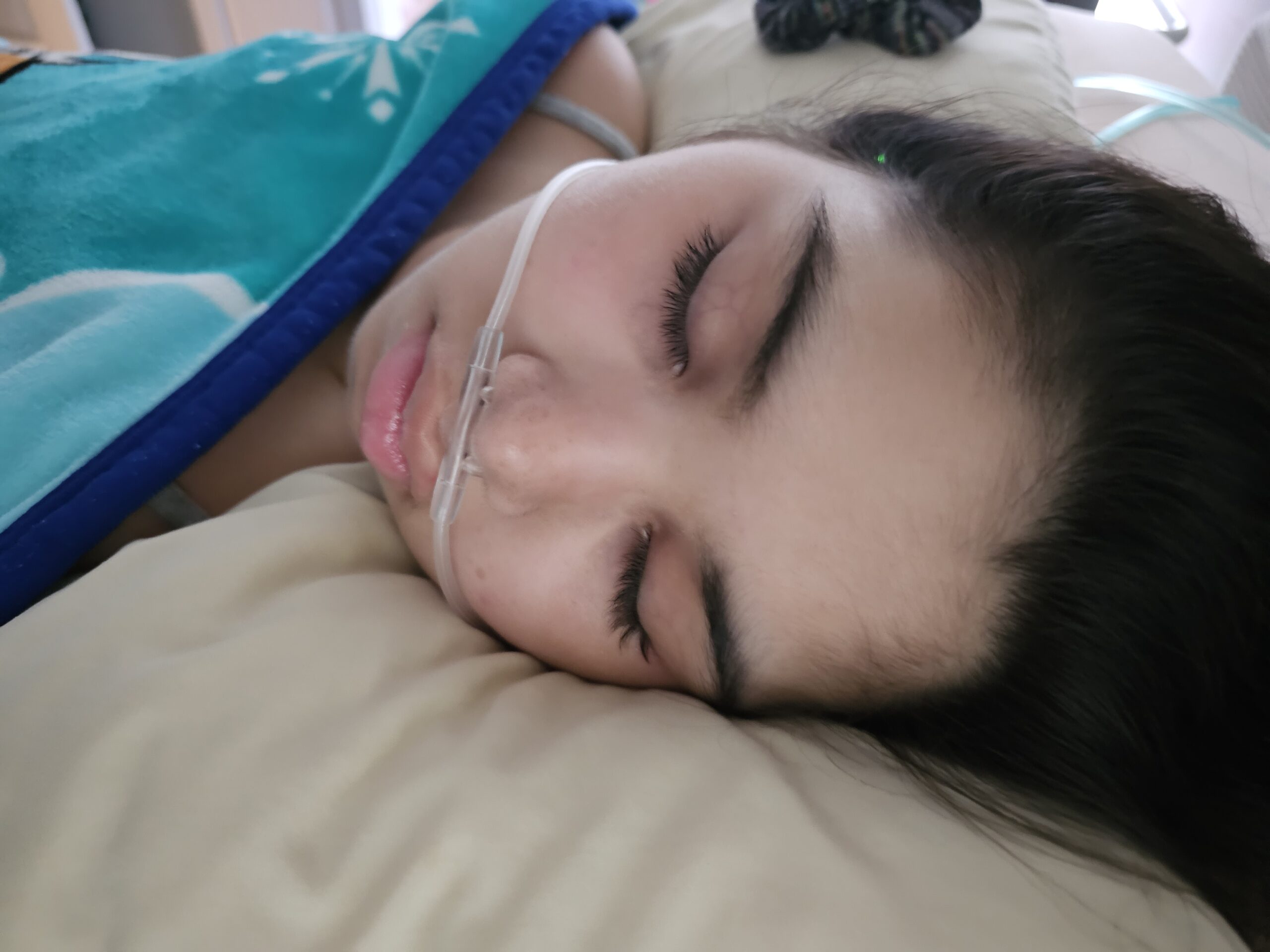
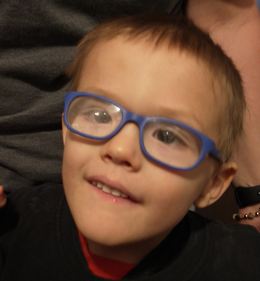
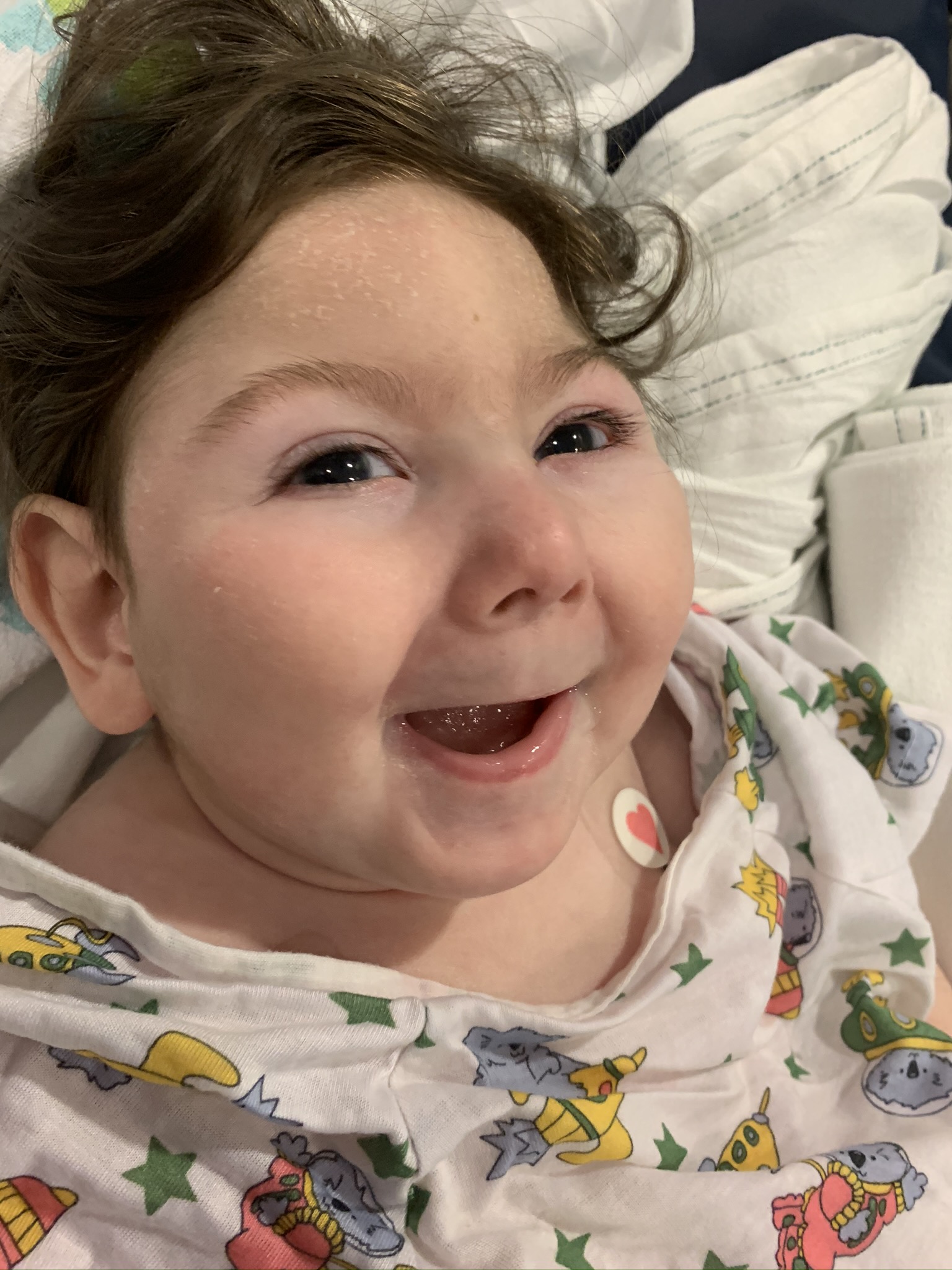
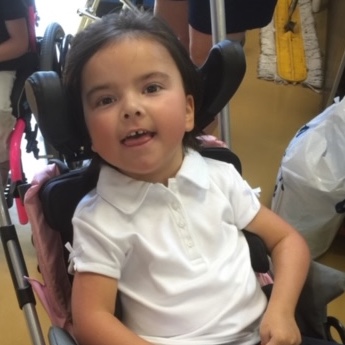
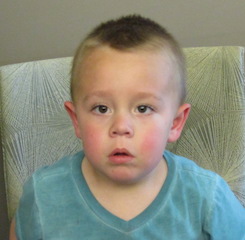
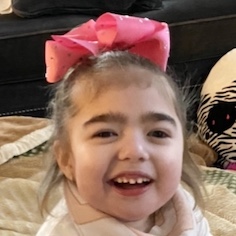
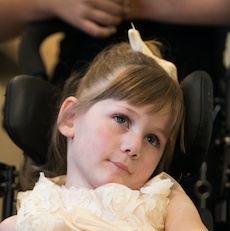
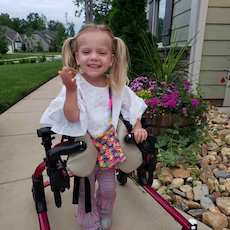
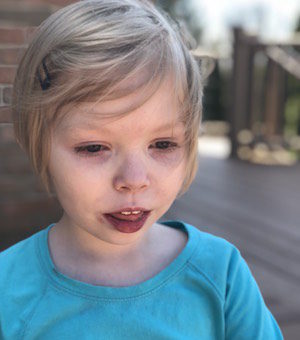
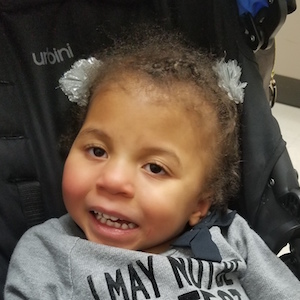
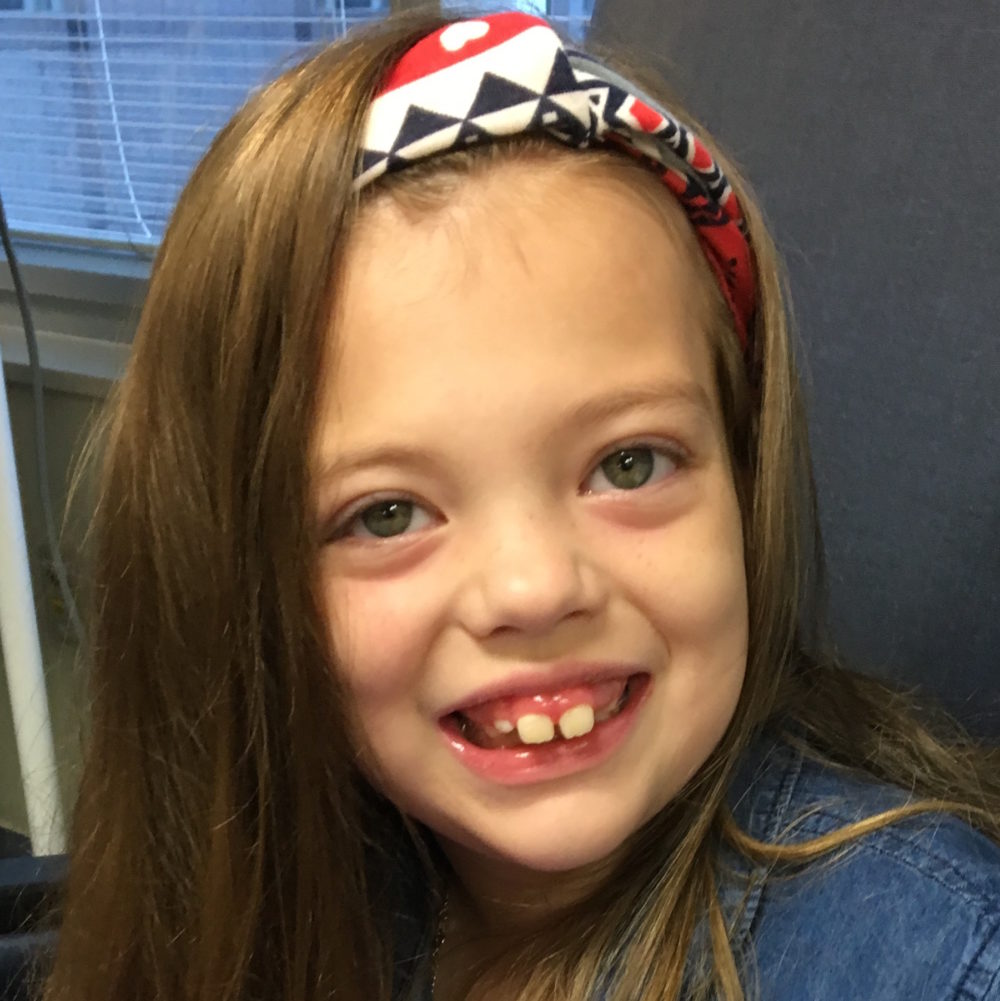
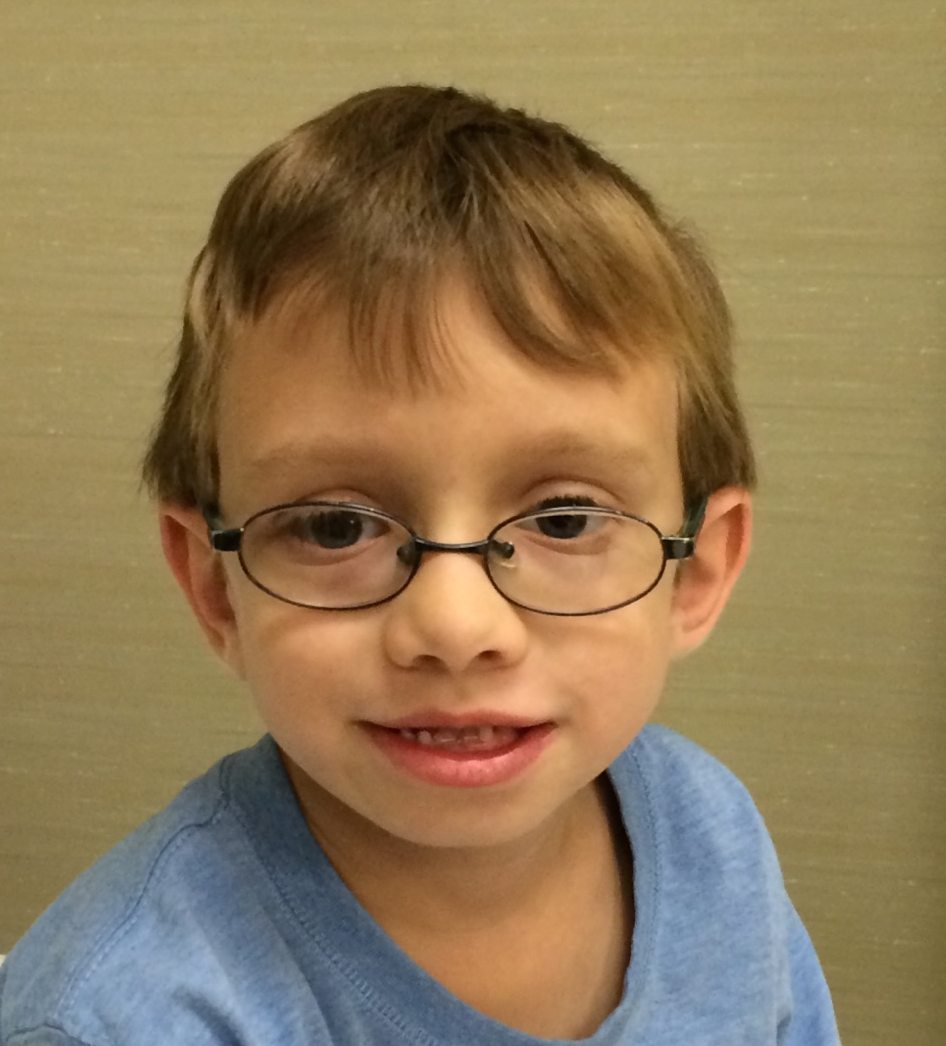
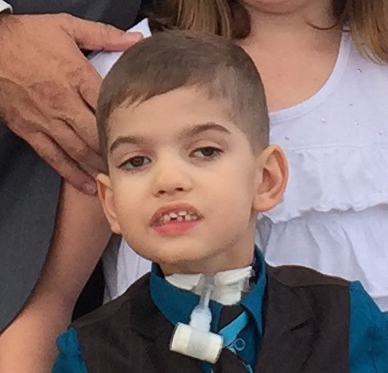
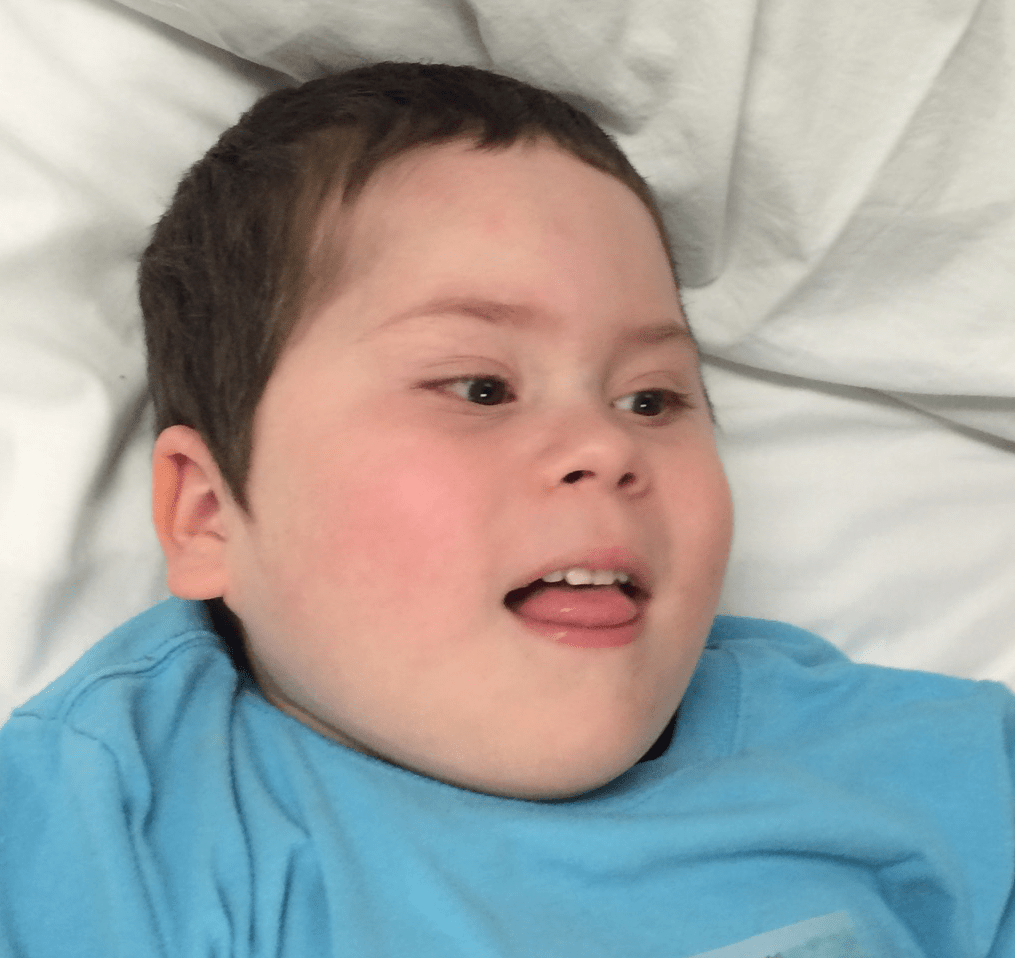
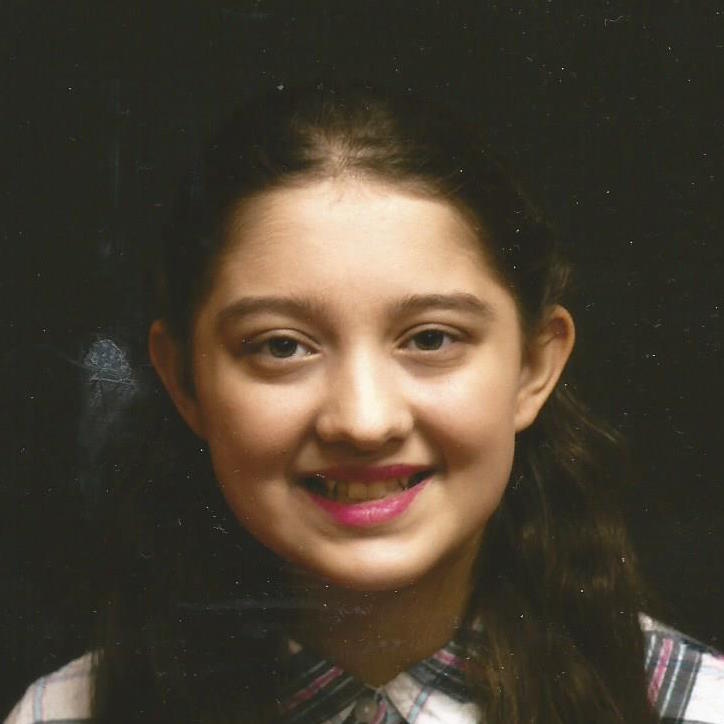Female, age 8, with hypotonia, delayed brain myelination, and developmental delays caused by a change in the CACNA1A gene
May 13, 2016
When the patient was 4 months old, she was noticed to have poor head control and low muscle tone. At 10 months, she had a brain MRI that showed mild thinning of the nerve fibers that join the two hemispheres of the brain (called the corpus callosum) and delay in myelination of deep white matter in the brain. At age 3, she had another brain MRI that showed a decrease in the area of the brain called the cerebellum. She does not have a history of seizures. Currently the patient is able to sit, crawl, and walk with the assistance of a walker. However, she does lose stability easily and primarily uses a wheelchair. She does not communicate verbally, but does communicate through sign language and an iPad. Overall the patient is delayed developmentally, but is making progress in gaining developmental skills and has not regressed in any skills. She feeds herself and is very social, engaged, and friendly.

Clinicians and researchers have identified the following genetic change to be causing the patient’s symptoms (Luo et al, 2017):
If this participant sounds like you or someone you know, please contact us!